
General
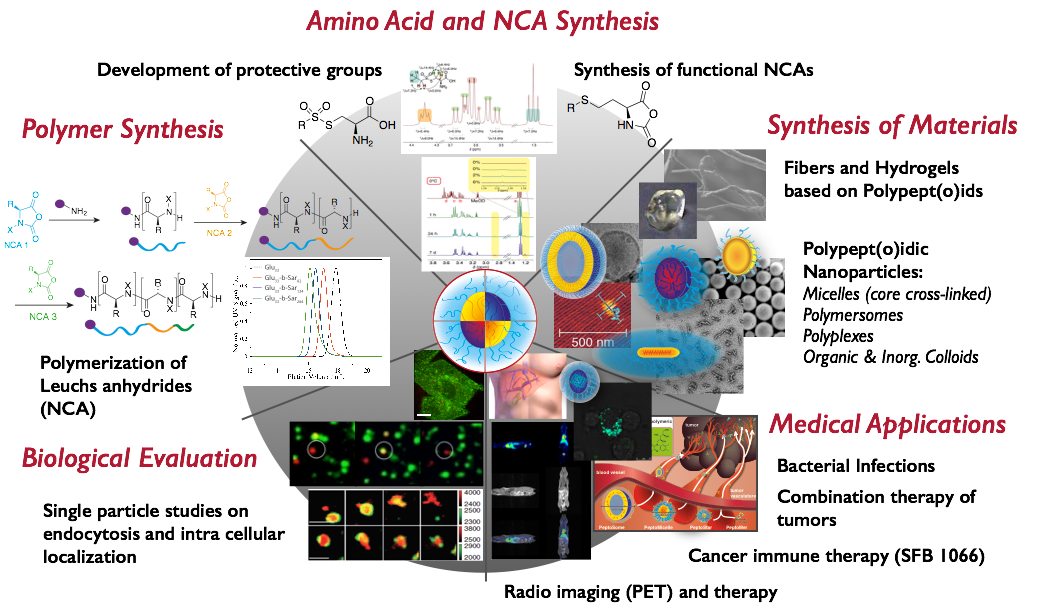
In our research, we focus on the development of polypept(o)ides – a hybrid material based on polypeptides and polypeptoids, e.g., polysarcosine. Our aim is to meet the complex requirements of therapeutic materials with robust and clean chemistry to develop not only potent therapies, but enable clinical translation. The reduction of synthetic efforts requires rethinking of existing concepts and developing new ones. With this in mind, we have developed protective groups for cysteines, which are stable against hard nucleophiles (ring-opening polymerization or solid phase synthesis), but reactive against soft ones (controlled disulfide formation), enabling post-polymerization of peptides by chemoselective disulfide formation. By applying these tools, we design nanoscopic or macroscopic materials based on polypept(o)ides either by chemical synthesis or by controlled self-assembly.
While we perform monomer, polymer and particle synthesis and characterization in-house (collaboration with Manfred Schmidt), we work in close cooperation with biologists, pharmacists and medical doctors to understand the biological behavior and to adjust our carrier systems to the biology of medical needs.
Development of Reactive Amino Acids and NCAs
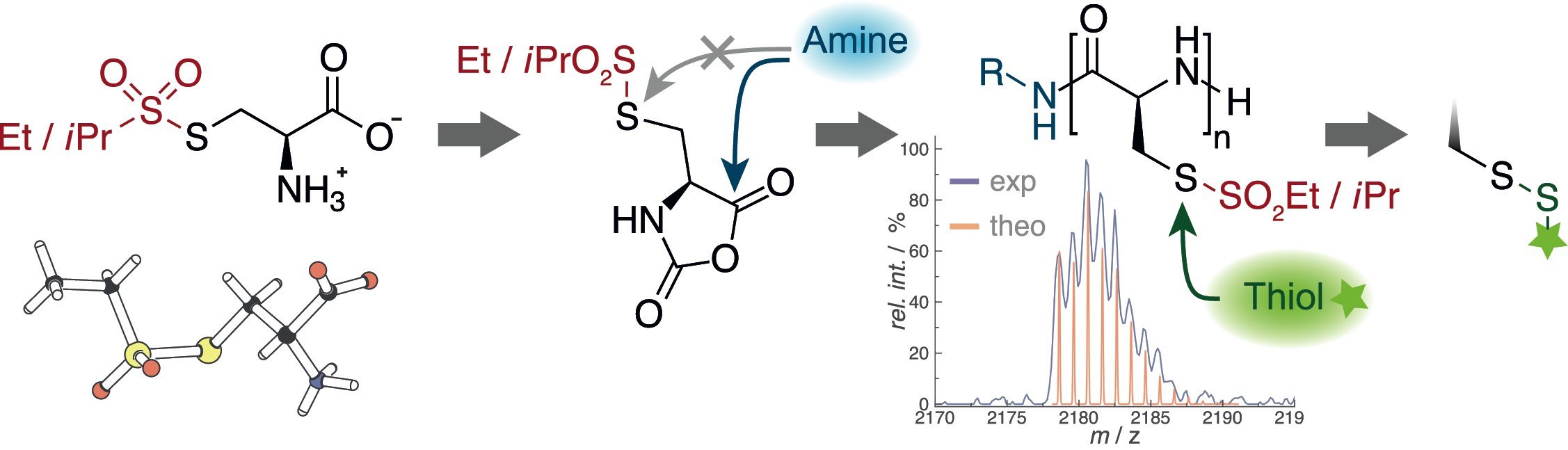
The first step in the development of multifunctional polypeptides is the synthesis of functional or reactive amino acids. Multifunctional polymers are often impossible to synthesize from a mixture of functional monomers due to problems in solubility, chemical group interferance with the polymerization itself, or simply due to differences in reactivity. On the other hand, standard solid phase (Fmoc cleavage) or NCA synthesis (acidic conditions) and polymerization (nucleophilic ring-opening polymerization by primary or secondary amines) expose functional groups to harsh reaction conditions. Thus, functional moieties are commonly protected (acids, thiols, alcohols and amines) and directly accessible reactive groups are limited, e.g. azides, alkynes or alkenes. Although these functionalities can be selectively addressed, they lead to C-C bond formation at the conjugation site, which are very unlikely to respond or to be cleaved in a biological environment. Therefore, we have developed a novel sulfonyl-based class of protective groups for cysteine and homocysteine, which are stable during peptide synthesis, NCA formation and polymerization but can be directly converted into disulfides. Disulfides are particularly interesting for biomedical appliactions since they are known to be stable in blood circulation, but are cleaved inside cells. The S-alkylsulfonyl groups (WO Patent 2015,169,908) are stable against primary and secondary amines, while they remain highly reactive towards thiols, yielding asymmetric disulfides in a quantitative manner within minutes at room temperature. Since the reaction of thiols with certain thiosulfonates proceeds in a highly selective manner and amines, alkenes or hydroxyl groups do not interfere, we can use this chemistry for site-selective conjugation of proteins or complex drugs in organic solvents or aqueous solutions.
Polymer Synthesis: Synthetic Polypept(o)ides
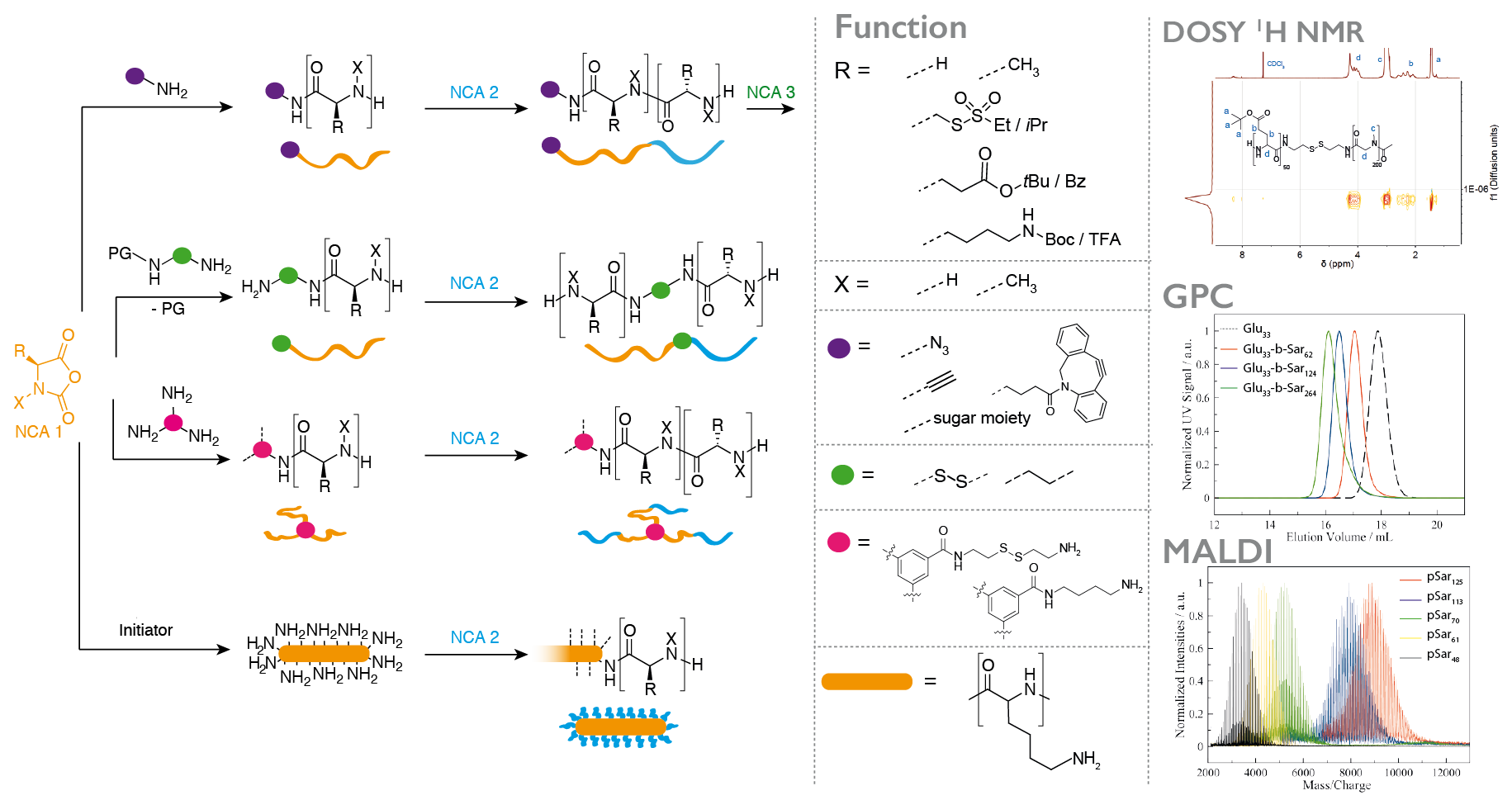
The application of polypeptides as biomaterials does often not require absolute sequence control, while a scalable synthesis and the incorporation of non-natural amino acids, e.g., N-substituted or reactive ones, is desirable. The ring-opening polymerization (ROP) of α-amino acid N-carboxyanhydrides (NCA) is therefore a versatile synthetic pathway to well-defined homo- and/or (block) copolypeptidic architectures. Whenever the polymerization itself is performed under controlled conditions, narrow molecular weight distributions, high end group integrities and low dispersity indices can be obtained. The NCA polymerization itself is not limited to natural amino acids, but versatile and scalable from milligrams to grams or even kilograms.
In most cases, we use polysarcosine (poly((N-methyl)glycine)) as a non-ionic, non-immunogenic and hydrophilic polymer in combination with α-amino acids since they can both be easily transformed into the corresponding NCA by the Fuchs-Farthing method and can be polymerized under living conditions, leading to polymers with high end group integrity and dispersities below 1.1.
In addition, the valence of initiators enables us to control the amount and position of functional groups within the polymer, control polymer branching and finally even adjust material properties by bottom up-synthesis.
Functional Materials: Controling Nanoparticle Properties by NCA Polymerization: PeptoStars, PeptoBrushes
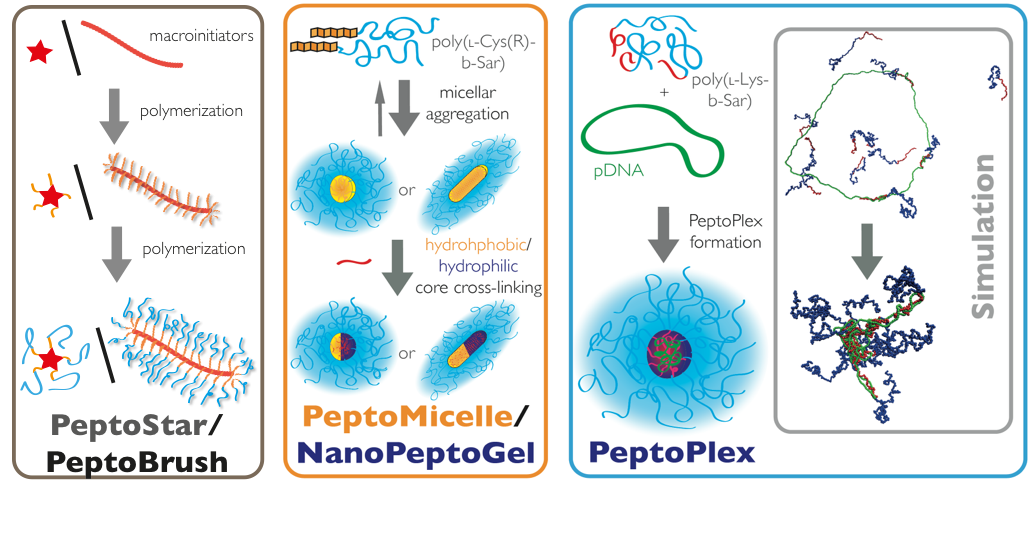
The most straightforward way to control nanoparticle properties such as size, shape, and internal structure is the synthesis of star-like or brush-like polymers. Oligovalent or multivalent initiators can be used to initiate the ROP, yielding spherical structures for small to medium sized initiators (cores containing 3, 6 or 12 amine functionalities) or worm-like for polymeric initiators (multivalent cores). The properties of nanostructures can be adjusted to the desired needs by controlling the block sequence and block length. After full conversion of the polymer block forming the hydrophilic corona (usually polysarcosine), the secondary amine end groups can be directly quenched with anhydrides or activated esters, introducing terminal functionalities. These functionalities can engage in post-polymerization modification reactions to attach bioactive moieties. In addition, the peptide blocks in the core of the system can be modified as well, which enables the synthesis of functional core-shell structures.
Secondary Structure-Controlled Self-Assembly: PeptoMicelles, NanoPeptoGels, PeptoSomes
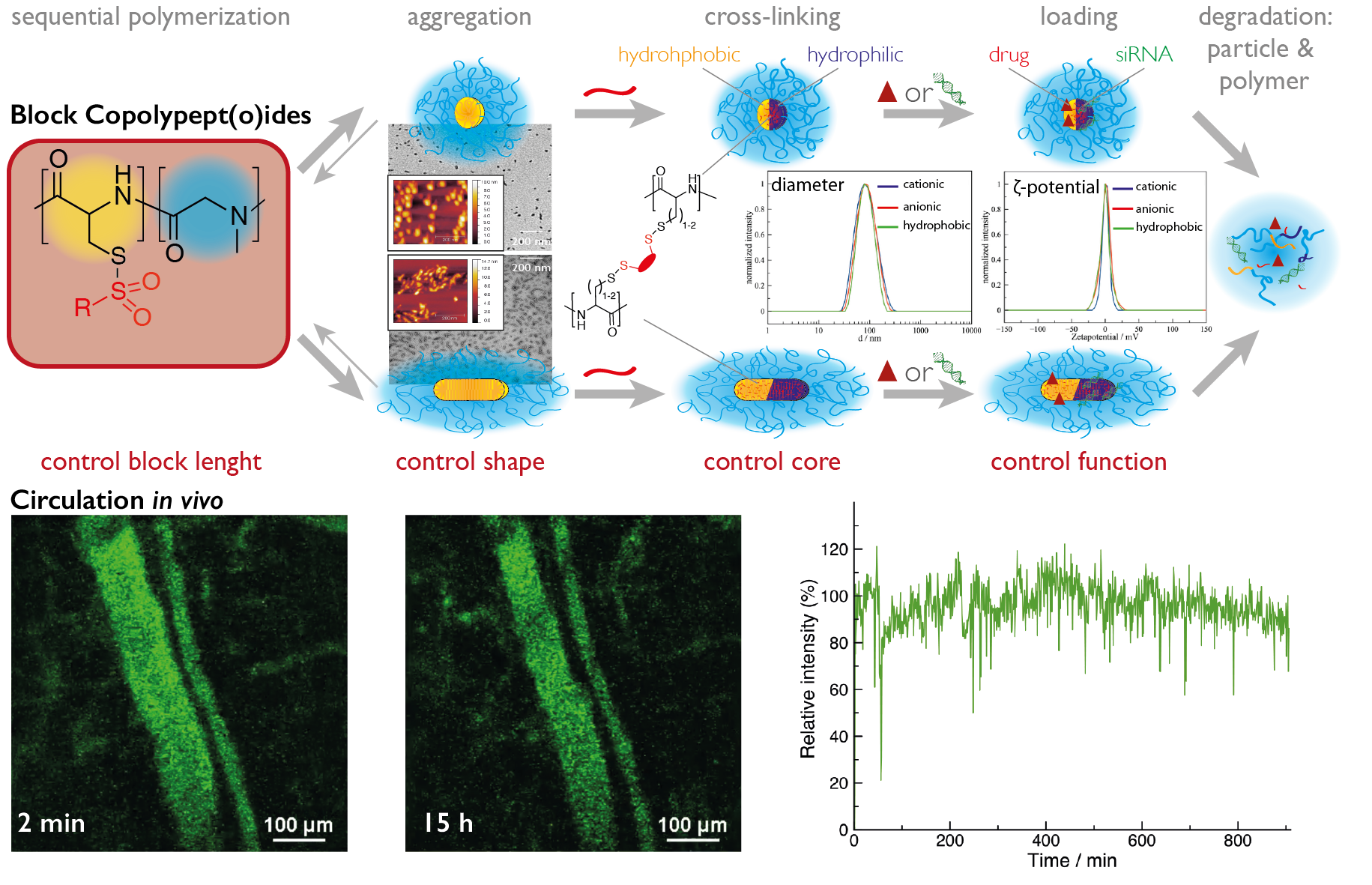
The most common way in the bottom-up synthesis of core-shell polymeric nanostructures is self-assembly. The control of this process is one of the major scientific challenges in our century since it opens up a versatile pathway to nanomaterials with enormous possibilities. While we employ electrostatic interactions for the formation of polyplexes by complexing pDNA or mRNA with a cationic block of a block copolypept(o)ide, we use hydrophobic interactions as driving force for the self-assembly of amphiphilic block copolypet(o)ides. These block copolypept(o)ides undergo spontaneous self-organization in aqueous solution (block-selective solvent) and can be used as polymeric surfactants to stabilize hydrophobic interfaces in aqueous media. We have successfully applied amphiphilic PSar-block-PGlu(OBzl) copolymers to emulsion polymerization, mini-emulsion techniques and in polymer-drug formulations.
Micelles, however, are of dynamic nature and in equilibrium with their unimers. Therefore, they possess reduced stability and are thus prone to premature disintegration and immune responses in vivo due to their amphiphilic character. Thus, stabilization of such aggregates appears necessary for most in vitro and especially for in vivo applications. Consequently, we can take advantage of the developed reactive thiosulfonyl-modified cysteines and homocysteines, which are hydrophobic and induce the formation of micelles in polar solvents (MeOH or water). Moreover, these polypeptides are highly reactive towards soft nucleophiles, enabling core cross-linking by disulfide formation with di- or oligo-thiols. Disulfide cross-linking is particularly interesting for biomedical appliactions since disulfides are known to be stable in blood circulation but cleaved inside cells due to a difference in redox potential (GSH concentration).
If the cross-linker of choice is hydrophobic, core cross-linked micelles result. On the other hand, the use of hydrophilic cross-linkers enables the synthesis of nanohydrogels. Based on this approach, we can use a given block copolypept(o)die and let it self-assemble in a block selective solvent into worm-like micelles (facilitated by β-sheet formation) or spherical micelles (suppressing of β-sheet formation by thiourea), multi-compartment micelles or vesicles and preserve the structure during cross-linking (see figure 6).
This pathway enables the synthesis of nanoparticle libraries that differ in morphology and core functionality. Interestingly, all nanoparticles, can be synthesized to have a neutral ζ-potential independent of their core polarity. Therefore, one single block copolymer can be used to synthesize either worm-like or spherical nanoparticles, where core properties can be decoupled from corona properties.
Template-Assisted Self-Assembly: PeptoPlexes
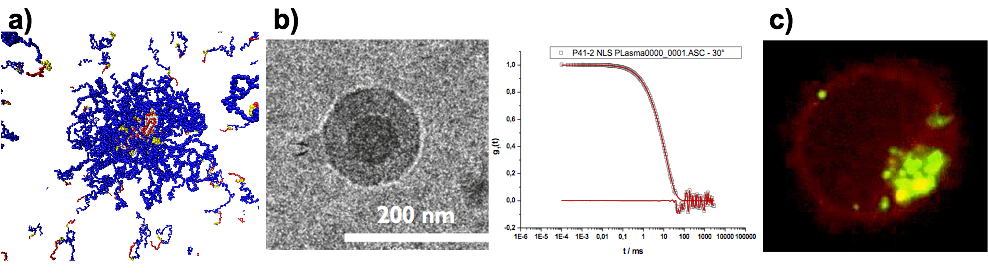
Besides secondary structure-controlled self-assembly, we are interested in polyplex formation between block ionomers and pDNA or mRNA. Block ionomers are block copolymers combining a cationic and a "stealth"-like block in a single double hydrophilic block copolymer. They can induce the formation of core-shell polyplexes, in which the cationic block complexes the nucleic acid, while the hydrophilic "stealth"-like block shields the assembly from undesired interactions with blood components and increases its stability towards disintegration. Furthermore, a third functionality is required to enhance transfection efficiency, which are endosomolytic groups, e.g. imidazole side chain of histidine, sequence-defined oligo(ethylene imine)s or viral peptides. These groups enable endosomal escape by membrane interactions or by the so-called proton sponge effect. Block ionomers for efficient transfection agents do not only require the combination of cationic/complexing, endosomolytic and "stealth"-like groups but also control over polymer microstructure. With a view towards a systemic application, polyplexes need to be stable in complex body fluids. Therefore, we incorporate the developed activated thiols (cysteine and homocysteine) as a middle block in the triblock copolymers. This novel type of block ionomer can complex nucleotides efficiently and allows for bioreversible stabilization by cross-linking using di- or oligo thiols after polyplex formation. We are currently screening synthetic and virus-derived endosomolytic cross-linkers leading to polyplexes, which are stable in serum, can be targeted to specific receptors and transfect the target cells more efficiently than jetPEI (poly(ethylene imine)-based transfection agent) or Fugene HD (lipid-based transfection agent).
